#2
Insulin-Like Growth Factor I
Protects Oligodendrocytes From Tumor Necrosis Factor- -Induced Injury -Induced Injury
Ye P, D'Ercole AJ
Endocrinology 1999 Jul;140(7):3063-72
The Univ of North Carolina at Chapel Hill, Dept of Pediatrics, Chapel Hill, NC 27599-7220, USA
PMID# 10385398; UI# 99312124
Abstract
Tumor Necrosis Factor-alpha (TNF-) has been causally implicated in several DeMyelinating Disorders, including Multiple Sclerosis.
Because Insulin-like Growth Factor I (IGF-I) is a potent stimulator of Myelination, we investigated whether it can protect Oligodendrocytes and Myelination from TNF--induced damage using mouse Glial cultures as a model.
Compared with controls, TNF- decreased Oligodendrocyte number by approximately 40% and doubled the number of Apoptotic Oligodendrocytes and their precursors.
Addition of Boc-Aspartyl(Ome)-Fluoromethyl ketone (BAF), an inhibitor of InterLeukin-1ß Converting Enzyme (ICE)/caspase proteases, blocked TNF--induced reductions in Oligodendrocytes.
Indicating, the TNF--induced reduction in Oligodendrocytes is, at least in part, due to Apoptosis, and that ICE/caspases are one of TNF- action mediators.
Simultaneous addition of IGF-I to TNF--treated cultures negated these TNF- effects nearly completely.
Furthermore, IGF-I promoted Oligodendrocyte precursor proliferation and/or differentiation in TNF--treated cultures.
To analyze TNF- and IGF-I actions on Oligodendrocyte function, we measured the abundance of messenger RNAs (mRNAs) for two major Myelin-specific proteins, Myelin Basic Protein (MBP) and ProteoLipid Protein (PLP).
While TNF- decreased MBP and PLP mRNA abundance by 5- to 6-fold, IGF-I abrogated TNF--induced reductions in a dose- and time-dependent manner.
The changes in MBP and PLP mRNA abundance could not be completely explained by the changes in Oligodendrocyte number, indicating that Myelin protein Gene expression is regulated by both TNF- and IGF-I.
These data support the hypothesis that TNF- can mediate Oligodendrocyte and Myelin damage, and indicate that IGF-I protects Oligodendrocytes from TNF- insults by blocking TNF--induced Apoptosis.
And, by promoting Oligodendrocyte and Precursor proliferation/differentiation and Myelin protein Gene expression.
#3
Myelin Oligodendrocyte Glycoprotein Induces EAE In The "Resistant" Brown Norway Rat
Disease Susceptibility Is Determined By MHC And MHC-Linked Effects On The B-Cell Response
Stefferl A, Brehm U, Storch M, Lambracht-Washington D, Bourquin C, Wonigeit K, Lassmann H, Linington C
J Immunol 1999 Jul 1;163(1):40-9
Max-Planck Institute for NeuroBiology, Dept of NeuroImmunology, Martinsried, Germany; Institute of Neurology, Univ of Vienna, Vienna, Austria; and Medical School Hanover, Clinic of Abdominal and Transplantation Surgery, Dept of Surgery
PMID# 10384097; UI# 99316285
Abstract
Experimental AutoImmune EncephaloMyelitis (EAE) induced by active immunization with the Myelin Oligodendrocyte Glycoprotein (MOG) is an Ab-mediated, T-Cell-dependent AutoImmune Disease that replicates the inflammatory DeMyelinating pathology of Multiple Sclerosis.
We report that disease susceptibility and severity are determined by MHC and MHC-linked effects on the MOG-specific B-Cell response that mediate severe clinical EAE in the EAE-resistant Brown Norway (BN) rat.
Immunization with the ExtraCellular domain of MOG in CFA induced fulminant clinical disease associated with widespread DeMyelination and with an inflammatory infiltrate containing large numbers of PolyMorphonuclear Cells and Eosinophils within 10 days of immunization.
To analyze the effects of the MHC (RT1 system) we compared BN (RT1 n) rats with Lewis (LEW) (RT1 l) and two reciprocal MHC congenic strains, LEW.1N (RT1n) and BN.1L (RT1 l).
This comparison revealed that disease severity and clinical course were strongly influenced by the MHC haplotype that modulated the Pathogenic MOG-specific AutoAntibody response.
The intra-MHC recombinant congenic strain LEW.1R38 demonstrated that Gene loci located both within the centromeric segment of the MHC containing classical Class I and Class II Genes and within the telomeric RT1.M region containing the MOG Gene are involved in determining Ab production and disease susceptibility.
This study indicates that the current T-Cell-centered interpretation of MHC-mediated effects on disease susceptibility must be re-assessed in Multiple Sclerosis and other AutoImmune Diseases in which AutoAntibody is involved in disease PathoGenesis.
#4
Interferon-ß-1b Treatment Modulates TNF And IFN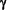 Spontaneous Gene Expression In MS Spontaneous Gene Expression In MS
Gayo A, Mozo L, Suarez A, Tunon A, Lahoz C, Gutierrez C
Neurology 1999 Jun 10;52(9):1764-70
Hospital Central de Asturias, Dept of Immunology, Universidad de Oviedo, Spain
PMID# 10371521; UI# 99297931
Abstract
Background
Interferon-beta (IFN-ß) lessens the overall frequency of acute attacks in patients with the Relapsing/Remitting form of Multiple Sclerosis (RRMS). IFN-ß may act by decreasing the synthesis of inflammatory Cytokines.
Objectives
To determine whether IFN-ß-1b treatment had an initial and sustained effect on the in vivo synthesis and secretion of Tumor Necrosis Factor alpha (TNF) and IFN.
Methods
A highly sensitive reverse-transcriptase PCR technique was used to measure baseline levels of mRNA in freshly isolated cells from patients before therapy and at 3, 6, and 12 months of treatment.
Also, protein concentration was measured in Serum and in culture supernatants from Mitogen-Stimulated Cells. The authors studied 16 patients, of whom 11 did not have clinical exacerbations, whereas 5 had one clinical relapse each during the study.
Results
Mean values of TNF mRNA levels in the 11 stable patients decreased significantly at 3 and 6 months of treatment in comparison with initial data.
After 6 months of therapy, IFN-ß-1b down modulated TNF transcripts in the 5 patients who experienced relapse.
In this group of patients, TNF levels rose sharply to reach pretreated values at 1 year of IFN-ß-1b treatment.
At the beginning of therapy, 6 patients had high concentrations of Serum TNF, which decreased to normal values following IFN-ß-1b therapy.
IFN mRNA expression also diminished after 6 and 12 months of IFN-ß-1b therapy in the group of stable patients, whereas nonrelevant variations were observed in patients who had one relapse.
Initially, patients' Peripheral MonoNuclear Cells secreted diminished amounts of TNF and IFN on PHA + PMA mitogen stimulation in comparison with normal control subjects.
After 1 year of therapy, IFN-ß-1b restored the normal production of TNF, whereas therapy did not restore IFN secretion to control values.
Conclusion
IFN-ß-1b decreases the spontaneous expression of two proinflammatory Cytokines.
#5
Allcock RJ, Christiansen FT, Price P
ImmunoGenetics 1999 Jun 8;49(7/8):660-665
Univ of Western Australia, Dept of BioChemistry, Nedlands 6009, Western Australia
PMID# 10369924
Abstract
Susceptibility to several disorders, including Insulin-dependent Diabetes Mellitus and Multiple Sclerosis, has been associated with alleles of HLA Class II Genes and loci in the TNF cluster in the central Major Histocompatibility Complex (MHC) region.
As recombination within this region is rare, it is difficult to determine which Genes are important. This will be facilitated by the identification of functional polymorphisms.
Hence we are sequencing reverse transcription-polymerase chain reaction products derived from central MHC Genes in well characterized and conserved ancestral haplotypes.
Here we address the IKBL gene, which lies near the TNF cluster at the telomeric end of the central MHC.
Although the IKBL cDNA sequence was conserved between most ancestral haplotypes, a synonymous nucleotide substitution, a 3' untranslated region substitution, and a single nonsynonymous substitution were identified.
The latter (IKBL+738) was present in multiple examples of the 7.1 haplotype [HLA-A3, B7, DR2, (DR15)] and resulted in a Cysteine to Arginine substitution in a predicted protein kinase C phosphorylation site.
This polymorphism did not occur in 18 other common haplotypes from the 10th International HistoCompatibility Workshop and thus appears haplospecific.
A role for IKBL+738 in the association between HLA-A3, B7, DR2, (DR15) and susceptibility to Multiple Sclerosis is discussed. |